


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
松本 淳
no journal, ,
In this study, we analyze the 3D-EM structures of smaller molecules bound to ribosome, such as tRNA and elongation factors, which are included in the EM density maps for the 70S ribosome by taking advantage of the best-fitting atomic models that were built in the previous study. One of the difficulties in this study is to spot the small molecules in the EM density map for the large 70S ribosome. This is solved by fitting the atomic model for the 70S ribosome into the EM density map and paying attentions to the regions where atomic model is not overlapped. In this way, we are able to isolate the 3D-EM structures that may correspond to the bound molecules. Our results show that the best-fitting atomic model of the 70S ribosome built in our previous study can isolate the regions for the bound molecules more effectively than the original PDB structure. We are especially interested in the bound tRNA molecules, and analyze the structures in detail.
石田 恒
no journal, ,
Ribosome is one of the supra-biomolecules used in the process of translating genetic information for the synthesis of polypeptides. In the course of its synthesis, two tRNA molecules move with mRNA through ribosome, changing their positions at the A (animoacyl), P (peptidyl), and E (exit) sites. This process, called translocation of tRNAs, is catalyzed by the elongation factor G (EF-G) using energy of GTP hydrolysis. Recent results from pre-steady-state kinetic analysis and cryo-electron microscopy (cryo-EM) suggest that there is a dynamic multistep process during translocation. However, the dynamic mechanism of translocation is unclear at the atomic level. We developed an EM density-fitting refinement method using all-atom molecular dynamics simulations including water molecules while retaining the condition that the constituent biomolecules fit in the EM density map. This method was applied to construct sequential states during the translocation using the X-ray crystallography structure of 70S ribosome complexed with EF-G corresponding to the post-translocational state (PDB code: 2WRJ) and two cryo-EM density maps corresponding to the translocational states (EMID code: 1365 and 1363). It was shown that multistep structural changes, such as ratchet-like motion between the small and large ribosomal subunits and head-rotation of the small ribosomal subunit, were observed during the translocation.
中川 洋
no journal, ,
タンパク質は生体内の機能分子である。生物はタンパク質によって機能を発現している。タンパク質は室温の水溶液中で機能している。これはタンパク質は熱揺らぎから逃れられないことを示しており、このようにタンパク質が揺らいでいることは現在広く認識されている。タンパク質の揺らぎは機能発現に重要である。タンパク質ダイナミクスを理解することは、揺らぎが生物機能とどのように関係しているかを明らかにするために必要である。中性子は、振動や拡散運動などのタンパク質ダイナミクスを定量的に調べる最も有効な測定手法の一つである。次世代中性子源J-PARCが茨城県に建設され、既に稼働している。この中性子源によって新たな生物物理学の展開が期待される。本発表では、タンパク質や水和水のダイナミクスについて紹介し、J-PARCでの生体物質ダイナミクス研究の展開を議論する。
村上 洋; 小野 正人
no journal, ,
生体高分子の水和水は生体高分子の機能に重要な役割を果たすと考えられている。蛋白質逆ミセルは実験的に水和水の状態を制御することができ、また、細胞中蛋白質のモデル系とみなせるため、蛋白質機能への水和水の効果や細胞中での生体高分子の機能の機構を明らかにするうえで、蛋白質逆ミセルの研究は重要と考えられる。本研究では、牛血清アルブミン蛋白質逆ミセル中に色素分子クマリンを導入し、その光化学反応の逆ミセルサイズ依存性をフェムト秒レーザーによる時間分解蛍光分光により調べた。フェムト秒からピコ秒領域はアップコンバージョン法を、ピコ秒からナノ秒領域では時間相関単一光子計数法を用いた。その結果、色素分子は逆ミセルを構成する界面活性剤(AOT)分子付近に局在していることがわかった。そして、水溶液中で見られるフェムト秒領域の超高速ダイナミクスが存在し、界面活性剤や界面活性剤に水和した水に起因するダイナミクス、そのダイナミクスへの蛋白質の効果や逆ミセルサイズ依存性を反応時定数及びエネルギー緩和量の観点から明らかにした。
徳久 淳師*; 甲斐 健師; 高 潤一郎*; 河野 秀俊; 郷 信広*
no journal, ,
XFEL can potentially offer a new mean for single biomolecule 3D-imaging. Measured intensity, which is a 2D diffraction pattern per one observation without phase information is extremely weak even using XFEL because sample is a single molecule in a single shot and incident intensity of XFEL cannot make stronger than a certain intensity due to suppress molecule decay. Under such experimental conditions, we consequently have to use extremely weak signals effectively to reconstruct a 3D molecule structure using multiply measured many 2D diffraction patterns. We have developed a two-step algorithm for reconstructing a 3D diffraction intensity data from many experimentally measured, quantum-noise limited 2D patterns of single molecule with unknown orientation. Once a 3D diffraction intensity data of a molecule obtained, the 3D structure can be obtained by applying oversampling method to the data. The developed algorithm enables us to detect as low as 0.1 photons per an effective pixel as signal for the classification in such a noisy experimental photon-count data. Theoretically, wave-number of a pixel giving such a limiting photon-count defines the attainable structural resolution.
樋口 真理子; Pinak, M.
no journal, ,
放射線により損傷が10から20ベースペア以内に複数の損傷が生じることがあり、これをクラスター損傷と呼ぶ。修復酵素hOGG1はDNA上の損傷残基である8オキソグアニンを修復する酵素だが、8オキソグアニンの相補鎖上の数ベースペア離れた位置にAPサイトや一本鎖鎖切断がある場合、修復が阻害されることが実験によりわかっている。8オキソグアニンと他の損傷の相対位置により修復率が変わるが、その原因は明確ではない。本研究では、クラスター損傷DNAと修復酵素hOGG1の分子動力学シミュレーションを行い、結合状態について調べた。クラスター損傷のモデルは8オキソグアニンと一本鎖鎖切断を含み、二つの損傷の相対位置を変えて、それぞれシミュレーションを行った。その結果、8オキソグアニンの隣のベースペアに鎖切断がある場合は、DNAが変形して結合面が増えるが、その一方でDNAの構造揺らぎが大幅に減少して、結合に不利に働くことがわかった。8オキソグアニンから3ベースペア離れた位置に鎖切断がある場合は、鎖切断の位置が酵素と接していないために影響が小さいことがわかった。
赤松 憲; 鹿園 直哉
no journal, ,
電離放射線によって生じたDNA損傷は、完全に修復されなければ突然変異や発癌の原因になるといわれている。特に、いわゆるクラスター損傷(複数の損傷がDNA上の狭い領域に集中的に生じている)は修復が困難とされているが、その実体はほとんど明らかになっていない。そこでわれわれは、このような仮説的な損傷を実験的に解明するために、蛍光共鳴エネルギー移動(FRET)等の蛍光消光現象を活用した損傷位置ばらつき評価法の開発を行っている。モデル実験として脱塩基部位(AP)を真ん中に有する31量体のDNAオリゴマー(相補鎖2本)を作成し、その各々にdonor蛍光分子(D), acceptor蛍光分子(A)を結合させた。D標識DNA溶液にA標識DNAを加えた後、D蛍光強度の経時変化を観察した結果、アニーリングが進むにつれてD蛍光強度が減少しA蛍光強度が増加する現象が観察された。このようなエネルギー移動現象はFRET以外にもさまざまな要因で生じることが知られており、それは主として蛍光分子の空間・時間分布を反映していると考えられる。本発表ではFRETを含めた他の蛍光消光現象についても考慮しながら、本メソドロジーの放射線照射DNAへの適用の実際について報告する。
 2によるインターロイキン13シグナルの阻害機構
2によるインターロイキン13シグナルの阻害機構松本 富美子; 畠中 孝彰*; 玉田 太郎; 本庄 栄二郎*; 太田 昭一郎*; 伊東 祐二*; 出原 賢治*; 黒木 良太
no journal, ,
アレルギー性疾患は喘息やアトピー性皮膚炎などに代表される炎症性の疾患で、近年劇的に増加しつつある。インターロイキン13(IL-13)はT細胞を介した免疫応答を引き起こす蛋白質であり、アレルギー疾患において重要な役割を果たしている。IL-13には2種類の異なった作用を持つ受容体に結合することが知られ、IL-13のIL-13R1あるいはIL-13R1/IL-4Rへの結合は細胞内シグナル伝達を促進し、IL-13R2への結合は伝達を抑制する。すなわちIL-13が関与する受容体群とIL-13との詳細な分子間相互作用の解析は、強力な抗アレルギー作用を有する新規有用分子の創製に重要な知見を与える。そこでわれわれは、カイコ-バキュロウイルス発現系を用いてIL-13にかかわる各受容体をFc融合体として発現・精製した。さらに、表面プラズモン共鳴(SPR)を用いてIL-13と各受容体との親和性を詳細に調べた。その結果、IL-13R2は、IL-4Rと相互作用することなく、IL-13との強力な親和性によってIL-13と結合してシグナル伝達を阻害していることを明らかにした。
金枝 直子; 石田 恒; 河野 秀俊
no journal, ,
ヌクレオソームは、ヒストンタンパク質とそれに巻きつくDNAから構成されており、真核生物のクロマチンの基本的な繰り返し構造である。DNAがヒストンに巻きついている構造のため、転写や遺伝子制御にかかわる結合タンパク質はDNAへの結合を阻害される。しかしタンパク質リモデラーは、DNAをゆるませて回転させる等DNAのポジショニングを変えることで、その阻害を取り除くと考えられている。このメカニズムを明らかにするため、DNAのポジショニングを変えた時の自由エネルギープロファイル計算を行っている。リモデラーの原子座標は、未だ決定されていないので、仮想的にリモデラーの役割を取り込む。その第一弾としてヌクレオソームDNAを仮想的にゆるませた状態を作り、ヒストンタンパク質を約10塩基対分だけ回転させて自由エネルギープロファイルを描く。結果として、自由エネルギーの最安定状態は、11度(約2, 3塩基対)で実現することがわかった。この結果は、一度DNAをゆるませると、リモデラーのようなDNAの位置変化を助けるタンパク質がなくとも、11度まで自発的に回転するということを示す。
 sp.593 (HaNDK)
sp.593 (HaNDK)新井 栄揮; 米澤 悌; 岡崎 伸生; 玉田 太郎; 徳永 廣子*; 石橋 松二郎*; 徳永 正雄*; 黒木 良太
no journal, ,
ヌクレオシドニリン酸キナーゼNDKは、あらゆる生物に保存された蛋白質であり、オリゴマー構造を形成することが知られている。グラム陰性細菌由来のNDKは基本2量体が2つ集まった4量体、真核生物及び古細菌由来のNDKは基本2量体が3つ集まった6量体などの会合構造をとる。本研究では、中度好塩菌由来HaNDKのE134A変異型の結晶構造を調べ、結晶構造中にグラム陰性菌由来MxNDKに類似した4量体構造と大腸菌由来EcNDKに類似した4量体構造が交互に現れることを明らかにした。この結果により、NDKが少ない変異導入によって多量体構造を容易に変化させることが示唆された。
米谷 佳晃; 河野 秀俊
no journal, ,
Molecular behavior of hydration water of DNA, in particular the DNA sequence dependence, has not been fully understood, in spite of many 3D complex structures determined by crystallography and NMR. Recently, we present that the sequence dependence of minor groove hydration is clearly correlated with that of DNA deformability based on the molecular dynamics analysis of 136 DNA sequences that differ from each other in their central tetramer [Yonetani, Kono, Biophys. J. 97, 1138, 2009]. To obtain an atomistic picture of the water dynamics and its DNA sequence dependence, we further calculated lifetimes of DNA-water hydrogen bonds. The results showed that the lifetime t varies from 1 ps to 300 ps, depending on the kind of basepair step. We found that the variation follows t = 1/kPm, where Pm is the probability of formation of multiple hydrogen bonds, which were often observed during the simulation. Based on this relation, we were able to explain the sequence dependence of lifetime in atomic view. Another topic we will present concerns DNA sequence recognition by DNA regulatory proteins. We are now performing molecular dynamics calculations to obtain free energy profiles for DNA-protein interactions. We will discuss how a DNA binding proteins pass over the non-target DNA and eventually recognize the target DNA.
米谷 佳晃; 河野 秀俊
no journal, ,
DNA deformability and hydration are both sequence-dependent and are essential in specific DNA sequence recognition by proteins. However, the relationship between the two is not well understood. Here, systematic molecular dynamics simulations of 136 DNA sequences that differ from each other in their central tetramer revealed that sequence dependence of hydration is clearly correlated with that of deformability. We show that this correlation can be illustrated by four typical cases. Most rigid basepair steps are highly likely to form an ordered hydration pattern composed of one water molecule forming a bridge between the bases of distinct strands, but a few exceptions favor another ordered hydration composed of two water molecules forming such a bridge. Steps with medium deformability can display both of these hydration patterns with frequent transition. Highly flexible steps do not have any stable hydration pattern. A detailed picture of this correlation demonstrates that motions of hydration water molecules and DNA bases are tightly coupled with each other at the atomic level. These results contribute to our understanding of the entropic contribution from water molecules in protein or drug binding and could be applied for the purpose of predicting binding sites.
増井 友美; 藤原 悟; 中川 洋; 片岡 幹雄
no journal, ,
トレハロースは生物乾燥耐性に重要な役割を果たしている。その重要な役割の一つに脂質を液晶相に保つことがある。従来研究は熱分析法を手法とし、トレハロースが乾燥状態での脂質のゲル・液晶相転移温度を下げることを明らかにし、トレハロースによる脂質膜の保護作用を明らかにしてきた。しかしながら熱分析法では試料調製法により脂質のゲル・液晶相転移温度が異なることが報告され、トレハロースがいかに脂質のゲル・液晶相転移温度を下げるのかは明らかにされていない。そこで、本研究では乾燥前の水が過剰に存在する溶液条件下で、小・広角X線散乱法を用いることによりトレハロースが脂質膜の構造に及ぼす影響を調べるとともに、中性子スピンエコー法を用いることでトレハロースが脂質膜のダイナミクスに及ぼす影響について調べることを目的とした。中性子スピンエコー法の結果、水溶液中でゲル相にある脂質膜はトレハロースの添加によって膜の曲げ弾性率が小さくなり、トレハロースの添加が温度上昇による効果と同一であることを明らかにした。
藤原 悟
no journal, ,
発表者がオーガナイザーを務める企画シンポジウム「Neutrons in Biologyの新たな夜明け; 大強度陽子加速器による中性子生物物理学の新展開」の第一講演として、中性子生物物理学の新たな可能性についての概観を行う。中性子と相補的なX線との比較を行い、中性子をプローブとした研究の特徴及びそのユニークさを示すとともに、中性子結晶学,中性子小角散乱,中性子非弾性散乱のそれぞれが、どのような可能性を持つかを紹介する。さらに、現在の原子炉JRR-3において生物学に対して利用可能な装置郡並びにJ-PARCにおいて運用中あるいは建設中の装置郡の紹介並びに仕様比較を行い、J-PARCが開く中性子生物物理の新たな可能性についての議論の出発点を与える。
藤原 悟; 米澤 康滋*; 松本 富美子; 小田 俊郎*; 武田 壮一*
no journal, ,
遺伝性心筋症は、心筋蛋白質の種々の変異により発症する。それらの変異蛋白質の中で、心筋収縮調節機構で重要な役割を果たすトロポニンの変異体に注目する。トロポニン(Tn)は3つのサブユニット(TnC, TnI, TnT)からなる蛋白質複合体である。トロポニンの種々の変異のうち、TnTのE244D変異及びK247R変異に注目する。これらの変異は、直接調節機能を担っている領域ではなく、Tn内の種々の調節領域をつなぐ領域に存在している。したがって、これらの変異がTn機能に及ぼす影響を調べることにより、心筋症発症機構のみならず、Tnの収縮調節機構そのものの解明につながりうると考えられる。これらのTn変異体研究の一環として、X線小角散乱法により、これらの変異体が溶液中でどのような構造をとっているかを調べた。その結果、変異体は、変異を持っていない通常のTnと同様にCa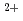 結合により構造変化が起こること、またTn変異体はTn野生体とは異なった構造をとることが明らかとなった。すなわち変異の導入によりTnの全体構造が変化することが示唆された。これはTnの変異により心筋症発症機構を考えるうえでの基礎をなす重要な結果である。
結合により構造変化が起こること、またTn変異体はTn野生体とは異なった構造をとることが明らかとなった。すなわち変異の導入によりTnの全体構造が変化することが示唆された。これはTnの変異により心筋症発症機構を考えるうえでの基礎をなす重要な結果である。
金枝 直子; 石田 恒; 河野 秀俊
no journal, ,
ヌクレオソームは真核生物に共通したクロマチンの基本的繰り返し構造で、ヒストンタンパク質とそれに巻き付くDNAから構成されている。細胞周期において、転写や複製,修復といった生命活動が起きるとき、ヌクレオソームDNAが大きく構造変化を起こすことがわかっている。この構造変化のメカニズムは、転写や複製,修復といった真核生物の生命活動の解明に手掛かりを与えてくれると考えられる。このDNAの構造変化が起きるときのメカニズムの解明に有用な方法の1つとして、自由エネルギープロファイルを描くことがあげられる。しかし反応座標の決定が難しい、計算コストが高いなどの理由から、現在のところ報告されていない。本研究はヌクレオソームDNAがゆるんだ構造をとり、ヒストンタンパク質の周りでスライドするときの自由エネルギープロファイルを、全原子モデルにより計算したものである。ヌクレオソームはヒストン修飾やDNAの配列の違い、ヒストンのアミノ酸配列のわずかな違いにより、遺伝子発現が制御されたりヌクレオソーム構造の安定性が違う等が実験的にわかっている。全原子モデルはこれらの構造の違いが取り入れられるため、エピジェネティクス研究に有用である。